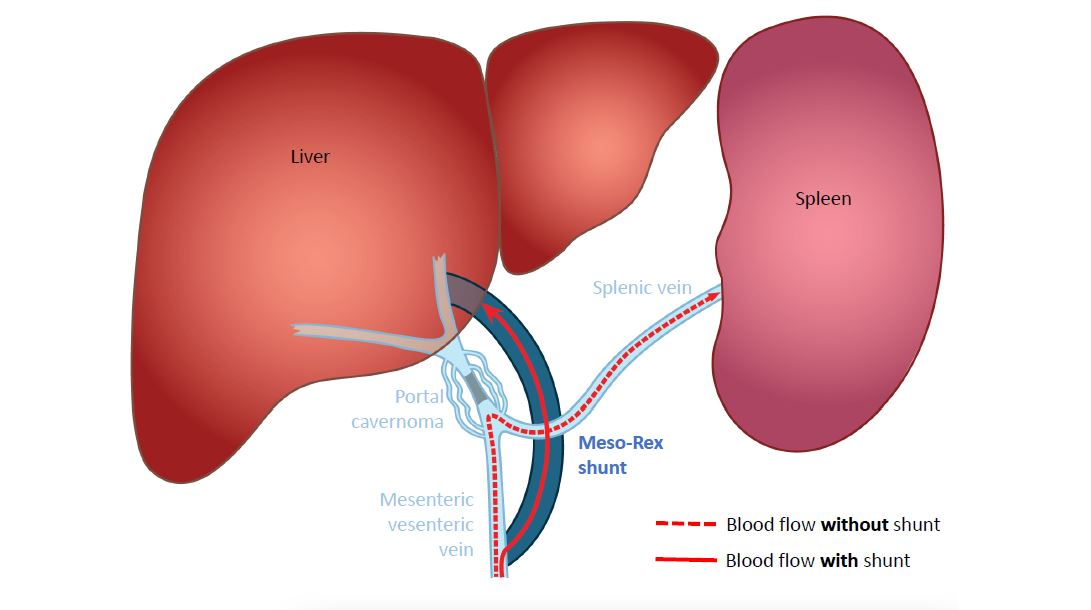
As mentioned in the video, the most common cause of portal hypertension in children is an obstruction of the portal vein outside the liver, called portal cavernoma. It can lead to bleeding esophageal varices or other problems. In this case, surgical treatment can be provided in most cases.
The video talks about a "shunt": this is the classic Rex shunt, also called the meso-Rex shunt. It is a kind of bridge to bypass the obstacle outside the liver, the portal cavernoma: the surgeons use a vein from the child's neck, which can easily be removed, and that will be placed between a large vein of the intestine, the superior mesenteric vein, and the Rex space, a location of the portal vein in the liver that is quite easily accessible by the surgeon.
In this way, the blood flow, blocked by the cavernoma and in hyperpressure, finds its way back easily through the bridge called the meso-Rex shunt, and can finally flow again normally, without resistance, through the liver. The physiological circuit is restored!